

作为持续深度跟踪药品监管政策的研究团队,我们在2015年9月份策略报告中,重点提示投资者应注意到本次药监部门注册审评政策改革“或许不一样”,而随着改革进程的推进,正逐步验证我们的观点。从监管部门系列政策动作看,药品注册改革已被定为CFDA最核心的改革任务:政策的重要性(本次注册改革已有3份国务院名义发布文件)、针对性(针对一致性评价从流程、技术原则等出台系列方案)、紧迫性(临床试验数据核查工作程序发布后第三天便公告首期核查计划)。而从CFDA监管层乃至国家更高层面领导的态度也表明,本次改革或许可能是历史性的。

第一次开展,并未持续推进。2012年1月20日,国务院发布《国家药品安全“十二五”规划》(国发[2012]5号)提出全面提高仿制药质量,分期分批开展仿制药一致性评价,尽管当时规划要求“国家基本药物目录、临床常用的仿制药在2015年前完成”。但直到2012年底, CFDA才由注册司发布《仿制药质量一致性评价工作方案(征求意见稿)》,随后2013年CFDA正式开始推进一致性评价,但因为各种原因推进进度缓慢,到2014年底,一致性评价工作基本陷入停滞状态。
第二次启动,高效迅猛执行。2015年8月18日,国务院发布《关于改革药品医疗器械审评审批制度的意见》(国发[2015]44号),文中明确指出加快仿制药质量一致性评价,力争2018年底前完成国家基本药物口服制剂与参比制剂质量一致性评价。而仅仅在2个月内,CFDA便由办公厅发布《参比制剂选择和确定》、《生物等效性研究技术》、《溶出曲线测定与比较》等核心技术方案的征求意见稿。高效迅猛的政策推进速度表明本次一致性评价或将显着区别于第一次。
国务院发文,仿制药一致性评价进入政策密集期。2016年3月5日,国务院办公厅正式印发《关于开展仿制药质量和疗效一致性评价的意见》(国办发[2016]8号)要求2007年10月1日前批准上市的化学药品仿制药口服固体制剂,应在2018年底前完成一致性评价,随着国务院文件的发布,仿制药一致性评价进入政策推动密集期。
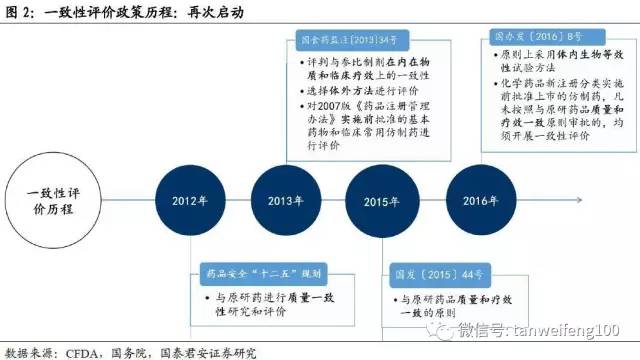
一致性评价工作涉及大量工作程序和技术性细节,包括参比制剂确定、整体工作程序、临床试验要求等,在国家整体框架确定后,CFDA以前所未有的高效率针对系列技术问题出台配套措施,需要特别强调的是本次一致性评价工作的整体协调已经由上一次的注册司调整为国家总局办公厅,再次表明本次一致性评价的重要性。
仿制药一致性评价是药品注册整体改革中的核心环节,在国务院发布《国务院关于改革药品医疗器械审评审批制度的意见》中推进仿制药质量一致性评价本身就是核心任务之一,而五大改革目标中除“提高审评审批透明度”外,目标均以一致性评价为基础。
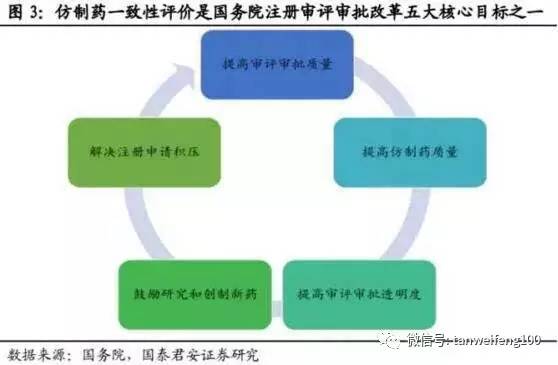
改革目标1-提高审评审批质量:仿制药一致性评价是一个系统工程,仿制药审评体系的建立可以有效完善CFDA审评架构,为建立更加科学、高效的药品审评审批体系奠定基础。
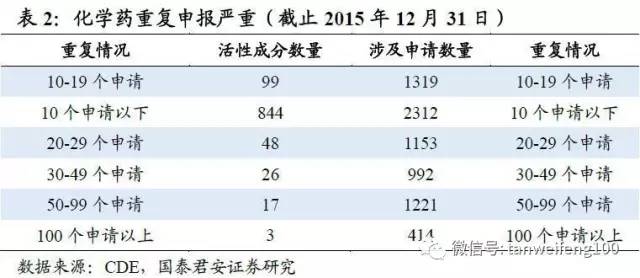
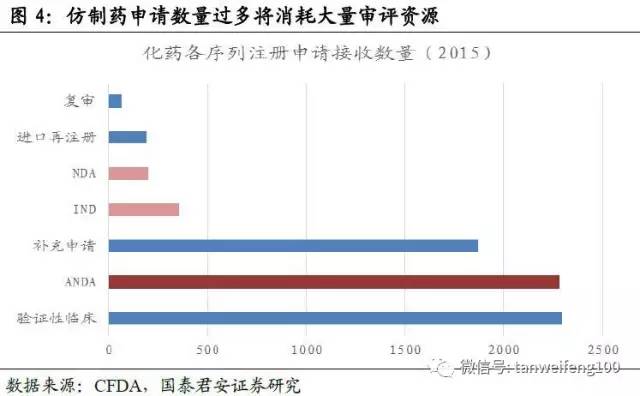
改革目标2-解决注册申请积压:截至2015年底,待审化药仿制药ANDA申请共7411个,占待审任务总量的42.9%,涉及活性成分1027个。重复申报较严重的有94个活性成分,涉及注册申请3780个,占化药ANDA总任务量的51%。本次一致性评价尽管只针对已有存量批文,但考虑到未来的监管压力以及药监部门对存量仿制药申请可能会采取类似于临床数据自查的清查模式,预计部分企业会选择撤回仿制药申请。
改革目标3-鼓励研究和创制新药:目前中国的药品申请结构中,仿制药申请依旧占主导,消耗大量注册审评资源,通过严格推行一致性评价,可以从源头引导企业改变经营策略,减少仿制药,尤其是重复性仿制药申报,将更多精力用于创新或者改良新药研发。
回顾过去近20年,CFDA的改革变迁过程,我们可以清晰地看到,药品注册审评一直是药品监管最为核心的工作,而无论是2005年《药品注册管理办法》在短短2年的时间里就进行修订,还是药品注册部门的人事变化,都让注册审评改革成为外界关注的焦点。
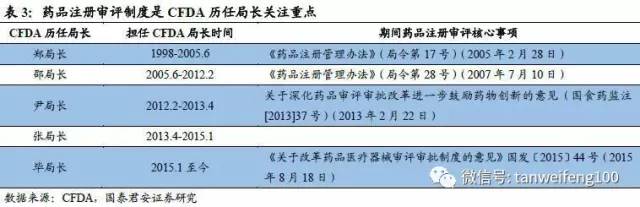
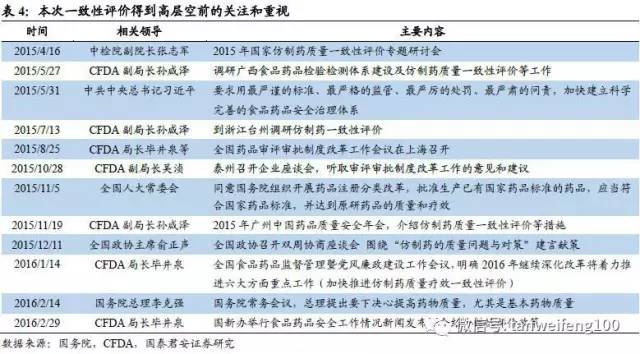
前文我们已分析一致性评价工作是药品监管注册改革的核心基础,从目前的情况看,可以认为仿制药一致性评价为CFDA领导层甚至更高层领导的核心工作,我们判断本次一致性评价可以定义为一场不能输的战斗。
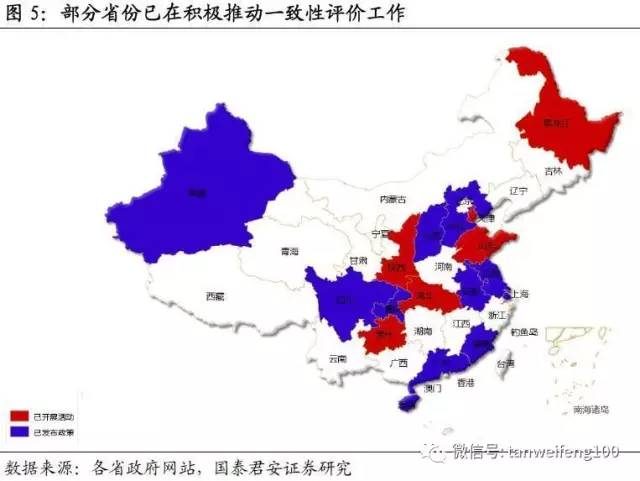
此外,自国务院发布一致性评价工作文件后,各省积极关注和推进仿制药一致性评价工作在地方开展。截至目前,湖北、陕西等多个省份通过工作座谈会或者业务培训等各种形式推进一致性评价工作落地。
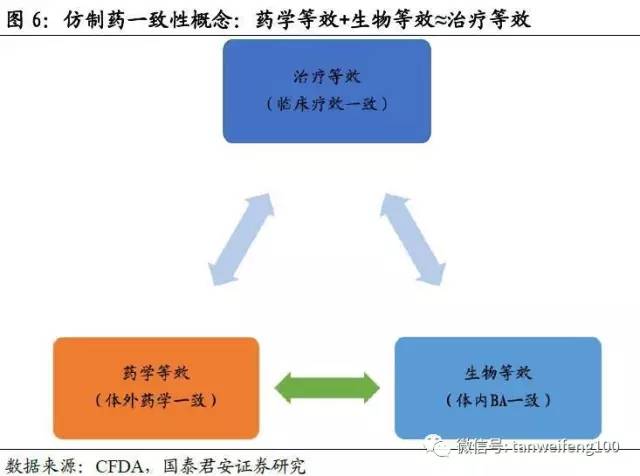
近些年国家对一致性评价标准也在逐渐变化,从早期的“质量一致性”到现在的“质量和疗效一致性”。而评价方法也从一开始的强调“体外评价方法”(体外溶出曲线)转变为现在的“体内生物等效试验方法”。对于一致性评价工作越来越严格,整体上强调仿制药的本质:即是原研药品的等效替代产品。
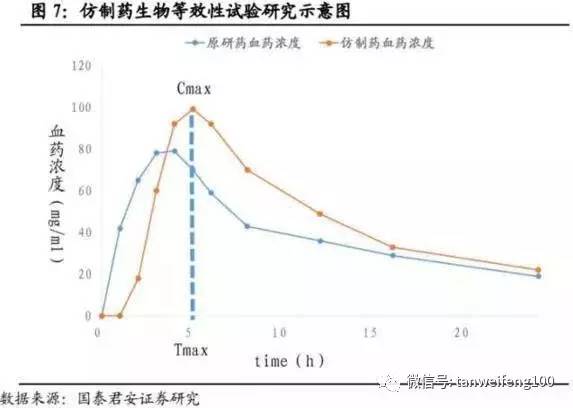
生物等效性定是指,在相似的试验条件下单次或多次给予相同剂量的试验药物后,受试制剂中药物的吸收速度和吸收程度与参比制剂的差异在可接受范围内(90%置信区间,标准值的80.00%-125.00%)。生物等效性研究方法按照研究方法评价效力,其优先顺序为药代动力学研究、药效动力学研究、临床研究和体外研究。如下图中的两条曲线为原药和仿制药的血药浓度曲线,Cmax/Tmax指达到最大血药浓度的时间。(此次评价关键指标Cmax AUC)

纵观美国和日本仿制药一致性评价发展历程,可以清晰看到,一致性评价并非一朝一夕可以完成,需要长时间的持续开展,美国到现在从首次开展至今有50年,日本从开展至今有45年,至今仍在进行。
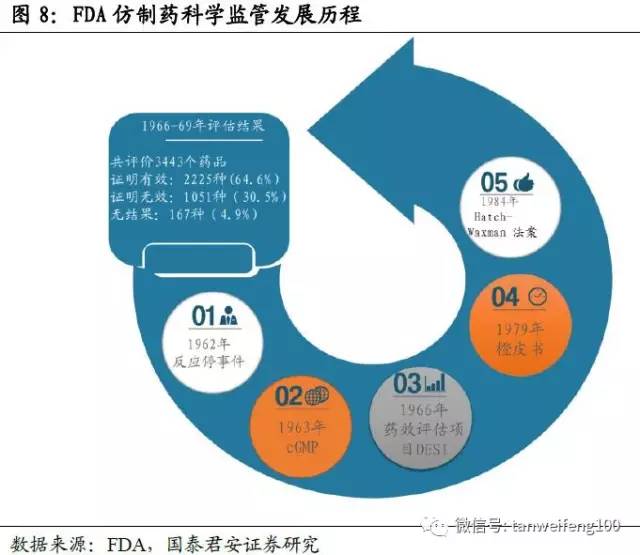
受“反应停”事件影响,美国于1966年首次开展了药品再评价项目,其评价的对象包括新药和类似药。美国药品一致性评价的核心思想是考察仿制药是否治疗等效于参比制剂,其中治疗等效包括药学等效和生物等效。
2004年1月起,FDA推出了“固体制剂溶出曲线数据库”,此后,溶出曲线的研究在药品一致性评价中发挥着越来越重要的作用。此处需要指出,不同于日本规定4条曲线的方法,美国仅选取了质量标准中规定的溶出试验条件下的一条溶出曲线。同时,FDA 也有一系列的相关指导原则分别对不同类型制剂、工艺放大、工艺变更等详细规定了溶出曲线.日本药品质量再评价:BE基础上的溶出曲线验证
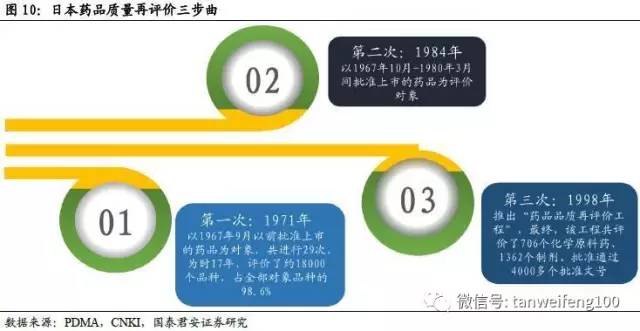
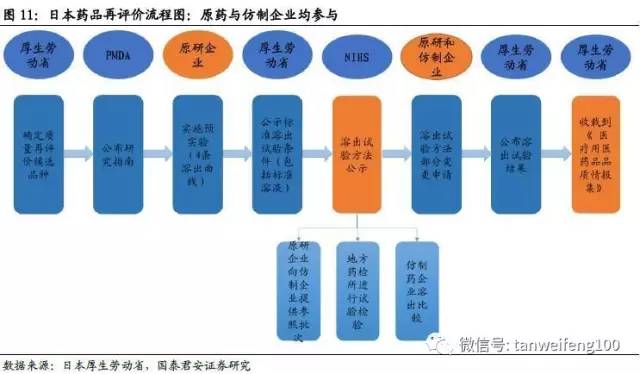
日本药品质量再评价的历史最早可追溯到1971年,纵观其发展演变历程我们可以看到,其质量再评价的内涵并不是单纯针对仿制药,评价对象也包括新药。核心理念是按照现阶段医学、药学等的学术水平,对已经批准的药品,从质量、有效性、安全性进行确认。分为对有效性、安全性进行再评价的药效再评价和对质量进行再评价(溶出)的质量再评价。
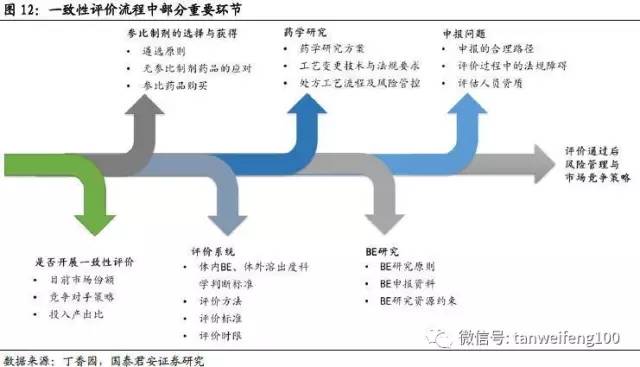
本轮一致性评价涉及的范围较大,可能会成为中国制药工业发展的分水岭,在具体的推进过程中可能会出现一系列的复杂情况,我们重点关注是否开展一致性评价、参比制剂的确定、临床资源的约束等三个核心问题。
根据政策要求,国家基本药物目录(2012年版)中2007年10月1日前批准上市的化学药品仿制药口服固体制剂,应在2018年底前完成一致性评价,其中需开展临床有效性试验和存在特殊情形的品种,应在2021年底前完成一致性评价;逾期未完成的,不予再注册。
按照业内对一致性评价工作整体流程和时间初步估算,完整的一致性评价需要24-30个月,企业开展工作的时间相当紧张。截止到4月8日,仿制药一致性评价工作程序的完整流程还未打通,即药品监管部门相关配套政策和审批流程尚未完善(尤其是参比制剂的选择和备案)。如果企业在一致性评价工作中某个环节遭遇挫折,则极有可能难以在国家规定的时间内完成评价工作,而与之对应,同品种中取得先发优势的企。




